GWAS AND ACCURACY OF PREDICTIONS FOR RESISTANCE AGAINST OsHV-1 IN PACIFIC OYSTER Crassostrea gigas
Introduction
Pacific oyster (Crassostrea gigas) is a highly important shellfish species accounting ~98% of global oyster production. Infectious diseases present a significant threat to the sustainability of aquaculture production with high economic losses due to mortalities caused by diseases. Ostreid herpesvirus (OsHV-1-µvar) originates highly contagious viral disease to Pacific oysters which is serious concern for oyster farmers globally. OsHV-1 may cause massive mortalities (~100%) especially in susceptible phase (larvae and small size oysters) . Selection and breeding for resistance against infectious diseases is highly valuable tool to help prevent or diminish disease outbreaks, and currently available advanced selection methods with the application of genomic information could pace up response to selection. Current study was designed to estimate genetic variation for resistance against OsHV-1, map quantitative trait loci (QTL), and explore potential of marker assisted and/or genomic selection
.
Material and Methods
This study comprises of a resource population of subsided 1520 offspring belonging to ~8 F2 -families produced from a cross of highly resistant grand-parents and susceptible lines (kept free of OsHV-1 infection since 2010). These offspring were selected from a large size population of ~16,000 offspring which experienced natural field outbreak of OsHV-1 in summer 2017. F2 individuals were recorded twice a day to check their status (alive vs. dead). Tissue sampling was performed for early dead individuals (i.e., mortalities in early phase of infection) and the late survivors (two months later). The tissue samples of F1 parents and grand-parents were also collected for further genomic work.
Additionally, the w hole genome re-sequencing (WGS) was performed on 67 individuals including grand-parents, F 1 parents and 5 individuals per F2 family (4 survivors and 1 dead) using Illumina HiSeq 2500 platform . Genotyping calling using sequence data produced a quality genotypes for 1,684,660 SNPs. The genotype data from the sequenced individuals (grand-parents, F1 parents and ~5 offspring per F2 family) was used for the construction of linkage map. Linkage mapping was performed with Lep-MAP3
using all the informative markers in VCF file.
The survival recorded F2 individuals (n~1520) were genotyped using 40,625 SNP array (Thermo-Fisher, AXIOM), and 14,245 markers remained after quality filtering. Out of 14,245 array markers, 14,218 were in common with the markers discovered from sequence data which were allocated linkage positions using map developed from sequenced individuals.
Estimates of genetic variation and the b reeding values were obtained using ASReml 4.0
implementing genomic or pedigree-based relationship matrices (G-matrix and A-matrix, respectively) with the following mixed model,
where is a vector of ‘n’ phenotypic records, is an overall mean, is a vector of additive genetic effects distributed as or is the incidence matrix for additive effects, and is the vector of random residual effects with . Accuracy of prediction was computed using a cross validation scheme by masking the phenotypes of ~ 20% of the offspring . The cross validation process was repeated 20 times and t he accuracy was computed as the correlation of the estimated breeding value (pedigree/genomic, PEBV/GEBV) with the phenotype scaled by the square root of heritability.
GWAS was performed by applying the same above model in GCTA program
with first 4 PCAs included as covariate and the SNP effects were also computed.
Results and Discussion
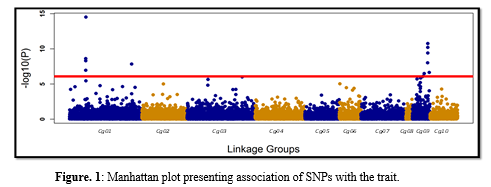
We developed a linkage map ~1.18 million markers grouped into 10 linkage groups (Cg ) which are consistent with the karyotype of this species. The estimated heritability for resistance to OsHV-1 (binary, dead/alive trait) using pedigree vs. genomic information was moderate to high (0.23 vs. 0.63, respectively). GWAS revealed strong signals of QTLs on two different linkage groups (Cg01 and Cg09, including 6 and 10 SNPs respectively), presenting significant association to the trait with P-value crossing genome-wide Bonferroni corrected threshold (Figure 1) . The proportion of total genetic variance explained by the top two SNPs (with single top-most SNP taken from each of the QTLs) was ~50% of the total genetic variance. The prediction accuracy for breeding values using genomic models exhibited 113% increase over the use of pedigree information.
In conclusion , results revealed moderate to high genetic variation for survival against OsHV-1 with two quantitative trait loci affecting resistance. The estimated breeding values using genomic information presented more than double accuracy compared to pedigree information, which should ultimately be reflected in overall genetic gain.
Acknowledgement
The research leading to the results of this study has received funding from EU-funded project VIVALDI (H2020 program, n°678589).
References
1. Meuwissen TH, Hayes BJ, Goddard ME: Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157(4):1819-1829.
2. Rastas P: Lep-MAP3: robust linkage mapping even for low-coverage whole genome sequencing data. Bioinformatics 2017(33):3726–3732.
3. Gilmour AR, Gogel BJ, Cullis BR, Welham SJ, Thompson R: ASReml User Guide Release 4.1 Structural Specification. In. Hemel Hempstead, UK: VSN International Ltd; 2015.
4. Yang J, S.H. Lee, M.E. Goddard, P.M. Visscher: GCTA: A tool for genome-wide complex trait analysis. Am J Hum Genet 2011, 88:76-82.