THE FIRST COMPLETE GENOME ASSEMBLY FOR THE MEAGRE Argyrosomus regius
Introduction
High-quality genome information is nowadays considered as a prerequisite of major significance in any animal and plant selection and breeding objectives. The meagre Argyrosomus regius is a fish species of elevated economic interest for the Mediterranean aquaculture in recent years, and there is ongoing effort to increase the efficiency of breeding performance. For this purpose, access to the full genomic sequence of the species would provide an important resource for exploring loci associated with quantitative traits and the genetic diversity of different wild populations and broodstocks. Here, we present the first complete nuclear genome for A. regius, which has been produced through a combination of long and short read technologies. This sequencing strategy, coupled with an efficient pipeline for assembling and polishing the genome, led to a contiguous, high-quality genome that provides an excellent base for future genomic studies in meagre.
Materials and Methods
Genome sequencing was carried out via a combination of third generation long read (Oxford Nanopore MinION) technology that provides contiguous long reads and a second generation (Illumina HiSeq4000) platform that produces short reads with high fidelity, which can be exploited to correct the former. Thus, a base assembly was first built using the Flye Assembler1 and polished from the long-read data, followed by polishing and error correction using the low error short read data (Racon2, Medaka (https://nellieangelova.github.io/De-Novo_Genome_Assembly_Pipelines/). Transcriptome sequencing was performed using total RNA extracted from 8 different tissues (listed in fig.1) on the Illumina HiSeq4000 platform. From these data, a final consensus transcriptome was constructed using Mikado7, incorporating intron junction, ORF prediction and homology information. Augustus8 and PASA9 were then used to carry out gene prediction based on the genome and transcriptome assemblies.
Results and Discussion
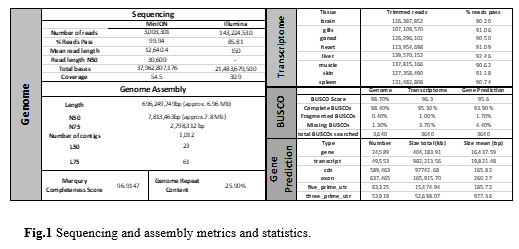
The MinION platform produced a total of 38,119,965,327 bp, with more than 99.5% passing quality control, giving a final 54.5 genome coverage and 3,003,301 sequences with a mean length of 12,640 bp. Illumina sequencing produced a total of 166,912,732 sequences of 150 bp, with more than 85.8% passing quality control, giving a 30.9 genome coverage. Transcriptome sequencing produced more than 100,000,000 sequences of 150 bp per tissue sample, with more than 90% of them passing quality control. The genome assembly produced from the long and short read data consists of 1,012 contigs, totalling 696,249,749 bp in size, with an N50 of 2,798,312 bp and an L50 of 23. This high level of contiguity is coupled with high completeness scores from both Merqury (96.9%, based on Illumina data kmer counting) and BUSCO (98.7%, based on conserved gene presence/absence), while the final consensus transcriptome also shows comparably high completeness (BUSCO 96.3%). Based on these data, the gene prediction pipeline annotated a total of 24,589 genes with a mean size of 16,437bp and a final BUSCO score of 95.6%. The constructed reference genome will set the basis for gaining deeper understanding of meagre biology and will boost the efforts for selective breeding through genomic selection.
Acknowledgments
The study has received funding from the Greek Republic through the "MeagreGen" project under the call "Special Actions – AQUACULTURE” in the Operational Program "Competitiveness Entrepreneurship and Innovation 2014-2020".
References
1. Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
2. Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
3. Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, (2014).
4. Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
5. Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
6. Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 1–29 (2020).
7. Venturini, L., Caim, S., Kaithakottil, G. G., Mapleson, D. L. & Swarbreck, D. Leveraging multiple transcriptome assembly methods for improved gene structure annotation. Gigascience 7, 1–15 (2018).
8. Stanke, M. & Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, 465–467 (2005).
9. Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).