DEVELOPMENT OF GENOMIC MARKERS ASSOCIATED TO PRODUCTION TRAITS IN LUMPFISH Cyclopterus lumpus
Introduction
Biological control of sea lice infection in Atlantic salmon cages has become an important alternative to tackle one of the most important diseases affecting salmon aquaculture. A handful of cleaner fish species have gained great importance in the control of sea lice (Barrett et al. 2020), among them, the use of lumpfish (Cyclopterus lumpus) has shown great promise (Imsland et al. 2018). Lumpfish is a sub-Arctic species found on both sides of the North Atlantic and commonly found along the Icelandic, Norwegian and British coastlines as well as the East coast of North America (Davenport 1985). Current lumpfish production is reliant on wild caught broodstock to meet the increasing demand. Lumpfish life cycle has been closed and hatchery reproduction is now possible.
Genomic resources are called to play a fundamental role in the improvement of selective breeding practices of aquaculture species. The development of these resources has been scarce for emerging species such as lumpfish, therefore, there is an imperative need to obtain genomic information that will support the establishment of effective and sustainable selective breeding programs. The recent release of the lumpfish genome sequence will facilitate the identification of genomic markers and the development of genomic tools. The aim of our study was to develop genomic tools and identify genomic regions linked to commercially important traits in lumpfish including gender and growth.
Materials and Methods
Fish used in this study belong to a population obtained from the Otter Ferry SeaFish farm (OFS). Ten lumpfish families from wild origin were produced in spring 2019 at OFS facilities. Four of these families were maintained in separate tanks until they reached a mean weight of 3-9g. Fin clips were obtained from all parents and from the four families including the 50 bigger, 50 smaller and 25 random fish from each family.
Two ddRAD libraries were prepared, containing all 536 samples and were sequenced on Illumina Novaseq 6000 platform. Reads were aligned against the genomic assembly of C. lumpus (NCBI Assembly accession GCA 009769545.1) using bwa v0.7.17 (Li & Durbin 2009) and assembled using Stack v2.41 (Rochette et al. 2019). Identified SNPs were collected from the 536 samples, and filtered according to the presence of at least two alleles, MAF >0.10, present in 75% samples and expected mendel segregation.
Genome wide association analysis was performed using the package R/SNPassoc v1.9-2 (González et al. 2007) using the “log-additive” model (except for sex, where ”co-dominant” model was used) and R/qtl2 v0.20 (Broman et al. 2019), analysing four traits, including gender, weight, condition factor and standard length.
Results and Discussion
Approximatelly 3.2 billion paired end reads were obtained as raw data from sequencing, from these, 3.2 million unique loci were detected and mapped across the available lumpish genome. After SNP calling and quality control, 10,630 informative SNPs were identified.
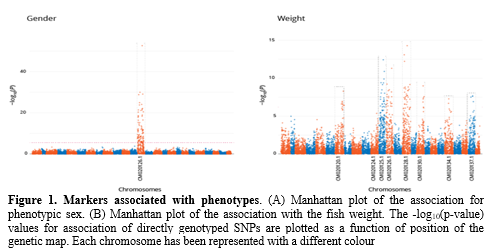
Association analyses were able to identify many genomic regions linked to the analyzed traits. The analysis of gender showed the highest association, identifying a single major QTL located in chromosome 13 (Figure 1A). Markers located in this region were further analyzed and a set of 16 markers was capable of accurately predict sex in all samples. Analysis of growh traits showed a polymorphic behaviour as expected, identifying significant association in many chromosomes, showing evidence of overlap in the QTL regions identified for weight and length in chromosomes 7, 12, 13, 15, 17 & 21 (Figure 1B). Of particular importance was the association between growth and gender shown in a region of chromosome 13 which could have implications in selective breeding. Moving forward, markers located in these identified QTL regions are candidates for the development of low density SNP panels that will provide a low cost alternative for producers to use genomic information in the development of selective breeding programs and improve production traits.
References
1. Barrett L.T., Overton K., Stien L.H., Oppedal F. & Dempster T. (2020) Effect of cleaner fish on sea lice in Norwegian salmon aquaculture: a national scale data analysis. International Journal for Parasitology 50, 787-96.
2. Broman K.W., Gatti D.M., Simecek P., Furlotte N.A., Prins P., Sen Ś., Yandell B.S. & Churchill G.A. (2019) R/qtl2: Software for Mapping Quantitative Trait Loci with High-Dimensional Data and Multiparent Populations. Genetics 211, 495-502.
3. Davenport J. (1985) Synopsis of biological data on the lumpsucker, Cyclopterus lumpus (Linnaeus, 1758). Food & Agriculture Org.
4. González J.R., Armengol L., Solé X., Guinó E., Mercader J.M., Estivill X. & Moreno V. (2007) SNPassoc: an R package to perform whole genome association studies. Bioinformatics 23, 654-5.
5. Imsland A.K.D., Hanssen A., Nytrø A.V., Reynolds P., Jonassen T.M., Hangstad T.A., Elvegård T.A., Urskog T.C. & Mikalsen B. (2018) It works! Lumpfish can significantly lower sea lice infestation in large-scale salmon farming. Biology Open 7.
6. Li H. & Durbin R. (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754-60.
7. Rochette N.C., Rivera-Colón A.G. & Catchen J.M. (2019) Stacks 2: Analytical methods for paired-end sequencing improve RADseq-based population genomics. Molecular Ecology 28, 4737-54.