LEVERAGING PUBLIC TRANSCRIPTOME DATA TO EXPLORE CELL TYPE AND TISSUE SPECIFIC EXPRESSION OF TARGET GENES IN SALMONID FISH
Introduction
Understanding the expression patterns of key regulatory genes is critical for elucidating their biological roles across different cell and tissue types. In fish species, such insights are especially important for advancing research in aquaculture, developmental biology, and environmental adaptation. Here, we leverage publicly available transcriptome datasets from the Gene Expression Omnibus (GEO) database to investigate both bulk and single-cell RNA sequencing (RNA-Seq) expression profiles of mucosa-associated lymphoid tissue lymphoma translocation protein (malt) gene orthologs in salmonid fish species, with a focus on rainbow trout (Oncorhynchus mykiss), a key salmonid representative . Utilizing a combination of bioinformatic pipelines, including transcript quantification, single-cell RNA-Seq methods, and differential expression analyses, we systematically mapped the expression landscapes of three malt paralogs present in rainbow trout across a range of tissues and cell types.
Material and methods
The workflow followed in this study is as follows (Figure 1):
Results and discussion
Initial quantitative RT-PCR validation revealed that malt1 expression is predominant in the head kidney of rainbow trout, while malt2 shows elevated levels in the spleen and trunk kidney. In contrast, malt3 exhibits higher expression in the liver and intestine compared to other tissues. These findings highlight a functional diversification of MALT paralogs that may reflect specialized immune and metabolic roles across different tissue environments.
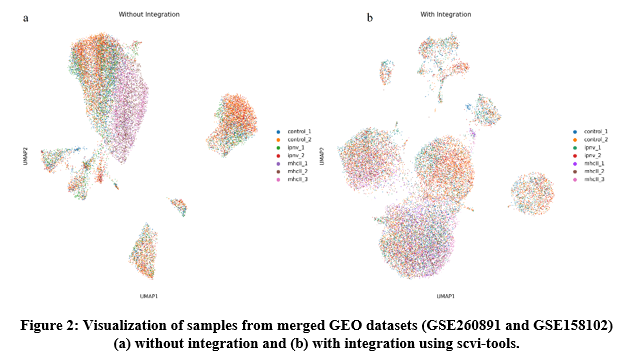
To further investigate these patterns, a meta-analysis combining bulk RNA-Seq and single-cell datasets is currently in progress. Integrating heterogeneous single-cell data, which encompasses diverse cell- type compositions, remains a considerable challenge. In this study, we demonstrate that the Single-cell Variational Inference (scvi ) framework offers a more effective strategy for harmonizing diverse single-cell datasets (Lopez et. Al, 2018) (Figure 2).
Our results reveal distinct, tissue-specific expression patterns of the three malt paralogs in rainbow trout. By integrating public transcriptomic resources with targeted experimental validation, our study underscores both the opportunities and challenges of functional genomics in aquatic organisms.
Furthermore, these insights lay the foundation for a deeper exploration into the regulatory mechanisms of immune and stress-response pathways in salmonids, with potential applications in fish health management and aquaculture improvement.
References
Edgar, R., Domrachev, M., & Lash, A. E. (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research, 30(1), 207–210. https://doi.org/10.1093/nar/30.1.207
Leinonen, R., Sugawara, H., & Shumway, M. (2011). The Sequence Read Archive. Nucleic Acids Research, 39, Issue suppl_1, D19–D21. https://doi.org/10.1093/nar/gkq1019
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I., & Yosef, N. (2018). Deep generative modeling for single-cell transcriptomics. Nature Methods, 15(12), 1053–1058. https://doi.org/10.1038/s41592-018-0229-2