THE CROSSTALK BETWEEN DIETARY RAYON MICROFIBERS AND THE FISH HOLOBIONT: IMPACTS ON GUT, SKIN, AND ENVIRONMENTAL MICROBIOTA
Introduction
Viscose-rayon microfibres (RFs) are polymers derived from the textile industry that are widely dispersed throughout aquatic environments worldwide. Whether ingested or simply present in the surrounding environment, their presence would affect both wild and farmed animals (Matias et al., 2024a ). Thus, a recent study on European sea bass (Dicentrarchus labrax ) showed that the increased presence of RFs in aquafeeds (CTRL – no RFs; RF1 – 0.001 g/kg; RF2 – 0.01 g/kg; RF3 – 0.1 g/kg) was linked to a n exponential increase in RF particles in the water, ranging from none in the tanks with CTRL-fed fish to 0.40 ± 0.21 RFs/L in those with fish fed the highest inclusion level (RF3) (Matias et al., 2024b ). This increased RF release and exposure was associated to a fatty liver without signs of growth impairment in the short term. However, a multi-tissue, multivariate gene expression analysis identified up to 19 discriminant genes, which depicted changes in hepatic lipogenic enzymes and intestinal/head kidney inflammatory markers, among others. Besides, these expression patterns denoted opposing regulation of immune responses at the systemic and local levels, which contributed to distinguishing CTRL_RF1 from RF2_RF3 fish. To further explore other disruptive physiological processes, this study aimed to assess the impact of RF exposure on the host and associated microbial communities, employing a multi-layered approach for the integrative profiling of gut, skin, and environmental water microbiota.
Materials and methods
European sea bass juveniles (5.8 g) were distributed in triplicate groups ( 180 fish per dietary treatment) in an flow through system. Fish were fed for 68 days to visual satiety with CTRL, RF1, RF2 and RF3 diets near to the visual satiety with automatic feeders (3 times per day) . At the end of trial , 9 overnight f asted fish per diet were randomly sampled for gut and skin microbiota. Water sample s (1L) w ere taken from the outlet water flow (OW) of each tank with fish, and from the inlet water flow (IW) from tanks without fish. Sequencing libraries were loaded in a MinION device , amplifying the full-length 16S rRNA gene with universal primers, using recently standardized sequencing procedures (Domingo-Bretón et al., 2024). Bi oinformatic analyses included demultiplexing, quality filtering and taxonomy assignment. Richness and alpha-diversity were calculated in R. Partial least squares-discriminant analysis (PLS-DA) w as used to depict the separation of experimental groups to be validated through permutation analyses (500 permutations). A minimum VIP value for the correct clustering of the 80% of samples was fixed in any microbiota layer, being 1.6, 1.3 and 1 the thresholds established for AI, skin and water, respectively . Bacteria taxa overcoming this threshold and displaying a high relative abundance (>0.5%) were selected as the main drivers. Inferred metagenome analysis was performed with Picrust2 tool and significant genes were considered at an FDR < 0.05.
Results and discussion
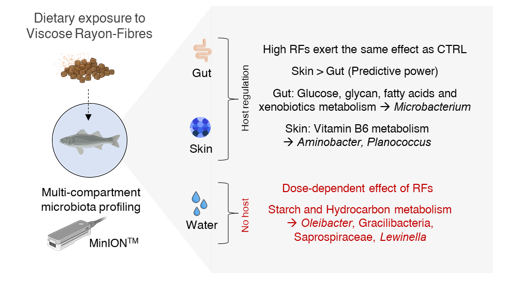
A total of 95,000 reads per sample were assigned to 978 taxa in at least one of the three microbial compartments. In both the gut and skin, dietary RF exposure decreased bacterial richness at low and intermediate doses (RF1, RF2), while similar values were found in CTRL and RF3 fish. In water samples, RF diets did not significantly alter microbiota richness, although the general trend was a decrease in bacterial diversity in RF2 and RF3 that reached similar values to those found in IW samples. Overall, these findings highlight that the presence of RFs in fish feeds can alter the microbiota of both the host and its surrounding aquatic environment, with different allosteric loads that seem to require a certain threshold level for activation. This assumption is corroborated by PLS-DA analysis, which clustered the AI and skin samples of the CTRL and RF3 groups together, while in water samples, the clustered groups were CTRL_RF1 and RF2_RF3. Discriminant analysis further revealed that the number of bacterial taxa with a discriminant value increased exponentially from AI (27 taxa) to skin (141) and water (792), again reflecting a different allosteric capacity – higher in animal mucosal environments than in the aquatic environment. Additionally, this buffering capacity appeared greater in the gut environment compared to the skin, the latter being more susceptible to environmental fluctuations due to its open nature. Upon applying an abundance filter, the number of discriminant bacteria was reduced to 3, 6, and 13 in AI, skin, and water, respectively. Inferred metagenomic analysis indicated that the abundant gut bacteria (Microbacterium and Alcaligenaceae spp.) are primarily involved in the activation of polysaccharide and xenobiotic metabolism, whereas predominant skin bacteria (Aminobacter and Planococcus ) are associated with vitamin B6 biosynthesis. In the water microbiota , up to 28 metabolic pathways appeared altered as a result of changes in taxa abundance , with particular relevance to starch and hydrocarbon metabolism through the action of Oleibacter , Gracilibacteria , Saprospiraceae , and Lewinella taxa.
Concluding remarks
This integrated perspective highlights the multifactorial nature of environmental pollution, microbial ecology, and host-microbe interactions, suggesting novel bacteria taxa and pathways through which microplastics exposure can influence host metabolic regulation.
Funding
This work was supported by the TNA programme (PID: 22708 and 25209) within H2020 AQUAEXCEL3.0 project (871108) to R. Matias for accessing to IATS-CSIC facilities.
References
Domingo-Bretón et al., 2024 . IJMS 25, 12603 ; Matias et al., 2024a . Sci . Tot. Env. 929, 172535 ; Matias et al., 2024b . AQUA2024, Copenhague, Denmark.