UNCOVERING THE FUNCTIONAL SYNERGIES OF AUTOCHTHONOUS AND ALLOCHTHONOUS GUT MICROBIOTA OF FARMED SEA BREAM
Introduction
The gut microbiota represents a complex environment inhabited by a large number of microorganisms that play an important role in numerous physiological processes of live animals, including fish (Singh et al ., 2025). Indeed, the microbiome present in the whole body, particularly in the gut systems of finfish and shellfish, not only contributes to digestion but also has an impact on nutrition, growth, reproduction, immune system , hereditary traits, and vulnerability of the host fish to diseases. Several factors can influence the gut microbiota structure and composition, including spatial distribution across the digestive tract and time post-feeding. Most microbiome studies target the colonizing autochthonous (resident or adherent) bacteria, as they are considered a mo re stable population with a more direct impact on the host . However, allochthonous (transient) bacteria may be also relevant with a recognized influence on resident bacteria and host physiology (Viver et al., 2023) . In any case, the organization of both communities, together with their functional role, still remains far from being clear . Accordingly, this study aimed to assess how the gut autochthonous and allochthonous bacterial communities change and interact in a spatial and temporal manner in farmed fish, using gilthead sea bream as a fish model of the Mediterranean aquaculture.
Material and methods
One -year old fish (356±12.8 g) were reared in 3,000 L flow-through tanks ( IATS aquaculture infrastructure) under natural photoperiod and temperature conditions in June with water [O2 ] levels maintained above 75% saturation. Fish were fed with a commercial diet (EFICO 3053, Biomar). At 24-h post-feeding , 10 animals were euthanized and samples of mucus and intestinal contents from the anterior and posterior intestine were collected for analysis of the autochthonous and allochthonous bacteria. Additionally, at 48-h post-feeding, mucus layer samples were taken from other 10 fish. Microbial DNA was extracted, and the 16S rRNA v1-v9 regions were sequenced (ONT MinION device) and processed with an in-house pipeline. Sequences were quality filtered and taxonomically assigned following a custom pipeline using SILVA database. A discriminant analysis (PLS-DA) of gut microbiota composition was made with EZInfo software. A Bayesian Network approach was conducted using t he SAMBA tool (Moroni et al., 2025). Clusters of nodes connected were identified using the Leiden community detection method . Functional profile was inferred for each cluster using the KEGG database for the pathways functional annotation protocol. Statistically significant differences between abundances and functions in resident and transient bacteria were assessed through a non-parametric Kruskal Wallis test.
Results and discussion
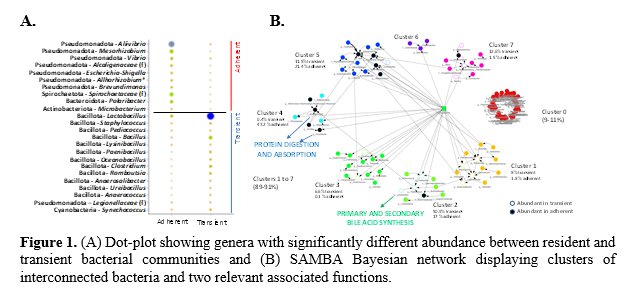
The transient bacteria presented greater values of richness (ACE), either in the anterior (286±83.3) or posterior intestine (276±40.6 ), in comparison to the gut adherent microbiota considered as a whole (71.2±3.29). Although the diversity indices did not reach a statistical significance , they shared a similar trend (e.g., Shannon index; transient bacteria, 2.09±0.147 ; resident bacteria, 1.44±0.138). Therefore, in agreement with previous studies (Sommer and Bäckhed, 2013), the mucus layer forms a more restrictive environment that supports the adherence and residence of a more stable microbial population . Besides, while the bacteria phylum Pseudomonadot a was dominant in the resident community (47-80%) , the Bacillota was the highest represented taxa in the transient microbiota (79-85%) . Certainly, the PLS-DA displayed a significant separation between resident and transient bacteria , confirming remarkable variations in their composition. Nevertheless, no clear or smaller differences were observed within each group between anterior and posterior intestinal bacterial communities , or across different post-feeding times. Thus, we focused on the comparison of resident and transient bacteria after 24-h post-feeding , regardless of the sampled intestinal section. At the genus level, m ost discriminant and abundant bacteria in the adherent community belonged to the phylum Pseudomonadota (e.g ., Aliivibrio, Mesorhizobium, Vibrio, Alcaligenaceae family, Escherichia-Shigella, Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium, Brevundimonas ) (Figure 1A) . Conversely, in the lumen layer, abundant bacteria were mostly of the phylum Bacillota (e.g ., Lactobacillus, Staphylococcus, Pediococcus, Bacillus, Lysinibacillus, Paenibacillus, Oceanobacillus, Clostridium, Romboutsia, Anaerosalibacter, Ureibacillus, Anaerococcus ). Numerous metabolic functions (147 ) were associated to both microbial communities , while 54 and 44 pathways were more represented in the resident and transient bacterial communities, respectively . Accordingly , the SAMBA tool allowed to discern different bacterial clusters (Figure 1B) that reveal different links and functional synergies between specific bacteria across the two microbial gut communities. F or instance, the Protein digestion and absorption appears likely linked , among others, to family Alcaligenaceae or the genus Polaribacter of the mucus layer. At the same time, t he Primary and Secondary bile acid synthesis became specially related to some lumen bacteria taxa, including several species of the genus Lactobacillus (R idlon et al., 2006).
Conclusions
Autochthonous and allochthonous bacteria form distinct communities in the gut of gilthead sea bream . Several genera inhabit both the mucus and lumen layer s, but most of them are differentially represented and interconnected . Thus, mucus-associated microorganisms appear essential to preserve gut function, including digestion processes, but at the same time bacteria of the lumen layer becomes necessary for the bile acid synthesis, likely modulating the composition of the mucosal bacteria community.
Funding
The work was funde d by MCIN NextGenerationEU (PRTR-C17.I1) and by Generalitat Valenciana (THINKINAZUL/2021/024), and BreamHOLOBIONT (PID2023-146990OB-I00) project.
References
Moroni et al. Microorganisms, 13:198 (2025 ); Ridlon et al. J Lip Res, 47:241 (2006); Singh et al. FEMS Microbio l Ecol , 101:fiae169 (2025 ); Sommer and Bäckhed . Nat Rev Microbiol, 11:227 (2013) ; Viver et al. Sci Total Environ, 889:164080 (2023).