ENHANCING BREEDING DECISIONS WITH COST-EFFECTIVE METHYLOME SCREENING: THE CASE STUDY OF MALE FERTILITY IN ARCTIC CHARR Salvelinus alpinus
Information
The epigenome is an unexplored resource in aquaculture breeding. Among the known epigenetic mechanisms, DNA methylation is the most well-studied, and it is suggested that it underpins several key traits in fish (Piferrer, 2023). Despite the substantial drop in sequencing costs over the last decade, screening the methylome in sufficient numbers as required by selective breeding practices remains challenging. By utilizing reduced representation sequencing methods, we can obtain genomic and methylome information in a cost-effective manner. As a proof of concept, we used reduced representation sequencing to screen the sperm methylome of Arctic charr (Salvelinus alpinus) males and investigated potential links to fertility as previously suggested (Pappas et al., 2025).
Materials and Methods
Fertility traits were recorded from 176 Arctic charr males from the Swedish breeding program. Sperm motility and kinematic parameters were measured using a Computer Assisted Sperm Analysis (CASA) system equipped with the SCA® Motility imaging software. Moreover, sperm concentration was counted using a NucleoCounter® SP-100™. Reduced representation sequencing libraries were prepared using methylation sensitive restriction enzymes. The library preparation followed a modified version of the original ddRAD protocol (Peterson et al., 2012) with previously documented adjustments (Franchini et al., 2017; Palaiokostas et al., 2015) , allowing at the same time scanning the methylome (Marconi et al., 2019). Sequencing was performed in one 10B lane of NovaSeq X Plus. Following sequencing and detection of single nucleotide polymorphisms (SNPs) using the Stacks software (Rochette et al., 2019), association studies for the recorded fertility traits were performed. Simultaneously, methylation levels were calculated and screened for putative associations with the male fertility traits using a weighted region comethylation network analysis with the R package comethyl (Mordaunt et al., 2022).
Results
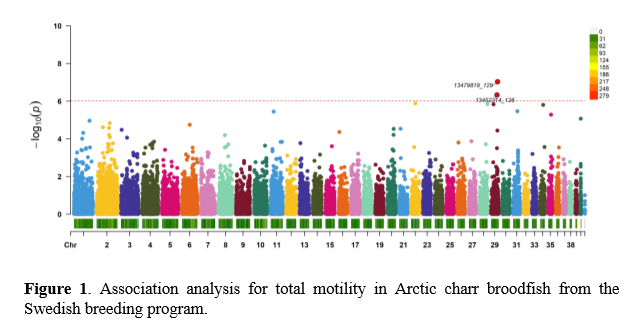
Approximately 15,500 SNPs satisfying quality control (found in at least 80% of the animals; minor allele frequency above 0.05; maximum heterozygosity of 0.6 and with a minimum minor allele count of 3) were retained for the association studies. Two SNPs located at chromosome 29 were found statistically significant at a genome-wide level (Figure 1; p < 0.05). In addition, 13 comethylated regions were constructed out of which one was found to have statistically significant association with sperm concentration (p < 0.05).
Discussion - conclusions
Using a cost-effective reduced representation protocol, we obtained genome- and methylome-wide information from over 170 Arctic charr males using one sequencing lane on an Illumina NovaSeq X Plus. As sequencing output continues to increase, our approach is expected to become even more cost-effective. Finally, we obtained novel information about the genome and methylome architecture controlling male fertility in Arctic charr.
References
Franchini, P., Monné Parera, D., Kautt, A.F., Meyer, A., 2017. quaddRAD: a new high-multiplexing and PCR duplicate removal ddRAD protocol produces novel evolutionary insights in a nonradiating cichlid lineage. Molecular Ecology 26, 2783–2795. https://doi.org/10.1111/mec.14077
Marconi, G., Capomaccio, S., Comino, C., Acquadro, A., Portis, E., Porceddu, A., Albertini, E., 2019. Methylation content sensitive enzyme ddRAD (MCSeEd): a reference-free, whole genome profiling system to address cytosine/adenine methylation changes. Scientific reports 9, 14864. https://doi.org/10.1038/s41598-019-51423-2
Mordaunt, C.E., Mouat, J.S., Schmidt, R.J., LaSalle, J.M., 2022. Comethyl: a network-based methylome approach to investigate the multivariate nature of health and disease. Briefings in Bioinformatics 23, bbab554. https://doi.org/10.1093/bib/bbab554
Palaiokostas, C., Bekaert, M., Khan, M., Taggart, J.B., Gharbi, K., McAndrew, B.J., Penman, D.J., 2015. A novel sex-determining QTL in Nile tilapia (Oreochromis niloticus). BMC Genomics 16, 171. https://doi.org/10.1186/s12864-015-1383-x
Pappas, F., Johnsson, M., Andersson, G., Debes, P.V., Palaiokostas, C., 2025. Sperm DNA methylation landscape and its links to male fertility in a non-model teleost using EM-seq. Heredity 1–13. https://doi.org/10.1038/s41437-025-00756-y
Peterson, B.K., Weber, J.N., Kay, E.H., Fisher, H.S., Hoekstra, H.E., 2012. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 7, e37135. https://doi.org/10.1371/journal.pone.0037135
Piferrer, F., 2023. Epigenetics in Aquaculture, in: Epigenetics in Aquaculture. John Wiley & Sons, Ltd, pp. 451–463. https://doi.org/10.1002/9781119821946.ch20
Rochette, N.C., Rivera‐Colón, A.G., Catchen, J.M., 2019. Stacks 2: Analytical methods for paired‐end sequencing improve RADseq‐based population genomics. Molecular Ecology 28, 4737–4754. https://doi.org/10.1111/mec.15253